改進(jìn)的QuEChERS-UPLC-MS/MS法測定動物源性食品巾13種全氟化合物(三)
發(fā)布時間:2021-05-30 16:14
編輯者:特邀作者周世紅
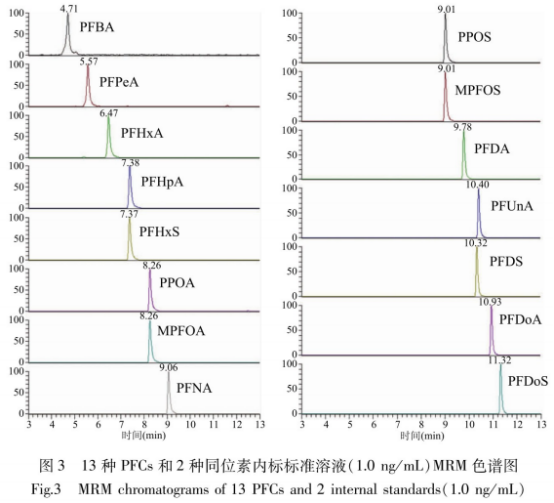
因此����,本實(shí)驗(yàn)采用AtlantisT3色譜柱,以2.5mmol/L乙酸銨甲醇溶液-2.5mmol/L乙酸銨水溶液進(jìn)行梯度洗脫����。圖3可見15種物質(zhì)在12min內(nèi)全部流出,日標(biāo)化合物保留時間適中���,峰面積�、信噪比較高����,雜峰響應(yīng)較小��;在空白基質(zhì)中添加日標(biāo)化合物,在上述檢測條件下�����,譜圖無干擾雜峰��,13種PFCs響應(yīng)好����。隨碳鏈的增加����,PFCs在C18柱上的保留逐漸增強(qiáng),同一碳鏈數(shù)全氟磺酸類化合物保留強(qiáng)于全氟羧酸類化合物����。PFBA最先流出,PFDoS最后流出����。
2.1.2質(zhì)譜條件選擇
PFCs化學(xué)結(jié)構(gòu)中具有羧基或磺酸基,因此采用負(fù)離子模式檢測����。本實(shí)驗(yàn)采用流動注射進(jìn)樣方式,分別對5μg/mL標(biāo)準(zhǔn)溶液進(jìn)行m/z200-1000ESI一級全掃捕,南實(shí)驗(yàn)結(jié)果發(fā)現(xiàn)�����,PFCs主要以電離后失去羥基上氫原子[M-H]-最強(qiáng)����,確定其為準(zhǔn)分子離子,并優(yōu)化噴霧電壓�����、蒸汽溫度���、離子傳輸管溫度以獲得較強(qiáng)的響應(yīng)值��。接下來�,以其作為母離子進(jìn)行子離子掃描并優(yōu)化碰撞能量�����,確定定性離子和定量離子�。在實(shí)驗(yàn)過程中,PFCAs生成[M-H-44]-子離子��,推斷是PFCAs發(fā)生中性丟失CO2產(chǎn)生;PFSAs生成m/z99和m/z80子離子����,推斷是PFSAs斷裂生成[FSO3]和[SO3]-。本次實(shí)驗(yàn)選是PFSAs斷裂生成[FSO3]和[SO3]�����。本次實(shí)驗(yàn)選取豐度較高且干擾較少的子離子[M-H-44]-和[SO3]-分別作為PFCAs和PFSAs的定量離子�����。
2.2前處理優(yōu)化
針對動物源性食品(肝臟����、腎臟��、肌肉)基質(zhì)復(fù)雜���、含有大量蛋白質(zhì)及內(nèi)源性物質(zhì)等特點(diǎn)����,本研究采用改良的QuEChERS樣品前處理方法�,酸化乙腈提取,C18、PSA和GCB混合吸附劑分散固相蒂取凈化��,減少基質(zhì)巾雜質(zhì)干擾��、提高回收率�����。
2.2.1提取溶劑優(yōu)化
已報(bào)道的文獻(xiàn)中采用乙腈����、鹽酸-乙腈作為提取溶劑提取動物源性食品的肌肉和肝臟基質(zhì)中的PFCs。鑒于乙腈是常用的動物源性食品有機(jī)提取溶劑��,而且PFCs是酸性化合物���,在酸性環(huán)境下呈非解離狀態(tài)��,有利于進(jìn)入有機(jī)相��,所以本實(shí)驗(yàn)選擇鹽酸-乙腈作為提取溶劑���。
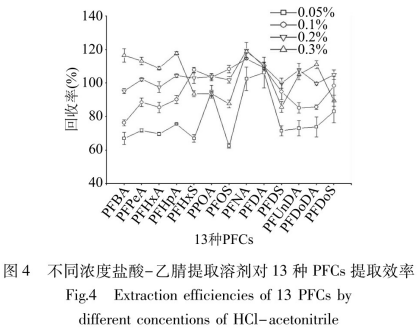
同時,考察了0.05%��、0.1%、0.2%�、0.3%網(wǎng)個不同濃度鹽酸-乙腈對動物源性食品中13種PFCs的提取同收率。圖4結(jié)果表明�,在0.05%~0.3%范同內(nèi),9種PFCAs的回收率基本是逐漸增加趨勢��,4種勢�,0.1%、0.2%����、0.3%鹽酸-乙腈的回收率均在合理范圍內(nèi)�����,分別為76.3%~114.6%����、95.3%~119.4%、85.8%~118.9%�����。上述三個濃度都是適宜的提取濃度�,但0.2%鹽酸-乙腈的回收率較集中�����,分布在95%~120%之問�����,最低回收率95.3%均高于其他兩個濃度����;同時為避免增加鹽酸用量對后續(xù)分析產(chǎn)生十?dāng)_�,所以選擇0.2%鹽酸-乙腈作為提取溶劑。在提取過程中�,0.2%鹽酸-乙腈使樣品基質(zhì)中大量蛋白質(zhì)變性形成沉淀,并通過高速離心去除����。但含脂量較高的樣品進(jìn)行蛋白沉淀時,易將樣品中的脂肪和水溶性雜質(zhì)也提取出來�����,造成提取液混濁��,可能對LC-MS/MS分析產(chǎn)生基質(zhì)干擾�,因此對提取液要進(jìn)一步凈化����。
2.2.2凈化方式優(yōu)化
QuEChERS方法是使用分散固相葦取技術(shù)凈化�����,通過將固相吸附劑直接加入到樣品提取液中來達(dá)到吸附干擾物質(zhì)的目的�����。動物源性食品中存在蛋白質(zhì)����、碳水化合物、色素�����、脂肪�、甾醇等物質(zhì)����,可以通過采用不同類型吸附劑達(dá)到凈化效果。已有文獻(xiàn)選用C18�、PSA��、GCB三種吸附劑單一或不同配比進(jìn)行PFCs凈化�,但存在樣品基質(zhì)種類較少或檢測項(xiàng)目少等缺點(diǎn)�����。本實(shí)驗(yàn)比較了3種吸附劑單一和混合方式對動物源性食品的肝臟����、腎臟和肌肉中多種PFCs的凈化效果及回收率。
2.2.2.1C18優(yōu)化
C18主要吸附脂肪和酯類等非極性共萃物���。通過在空白樣品中添加目標(biāo)物���,做2μg/kg添加濃度3個平行,比較40~100mg單一吸附劑C18對PFCs回收率的影響�����,發(fā)現(xiàn)隨著C18用量由40mg增加到80mg�,同收率亦增加,在80mg達(dá)到88.4%~116.3%(見圖5)�����;由80mg增加到100mg,大多數(shù)PFCAs回收率基本保持不變��,4種PFSAs和PFHpA�����、PFDA回收率有所下降�,但13種PFCs回收率仍處于85.5%~118.0%的合理范圍(見圖5)。因此采用合理同收率的最小用量80mg���。但凈化后的溶液有色素殘留����,會污染色涪柱及檢測儀器��,需要進(jìn)一步去除色素��。

2.2.2.2PSA優(yōu)化
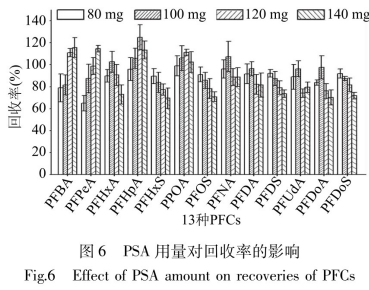
PSA是一種弱陰離子交換劑�����,可以吸附碳水化合物�、有機(jī)酸�����、少量色素等極性干擾物質(zhì)。在空白樣品中添加日標(biāo)物�����,做2μg/kg添加濃度3個平行����,在80mgC18凈化基礎(chǔ)上,同時加入80~140mgPSA觀察PFCs的叫收率變化情況��。由圖6可以得知隨著PSA用量的增加�,PFCAs回收率呈先上升后下降趨勢,并在100�、120mg時回收率達(dá)到合理最大值;PFSAs同收率呈下降趨勢��,80�����、100mg同收率是83.9%~92.1%�����。綜合考慮,選擇100mgPSA為最佳用量�。但凈化后的溶液有色素殘留,需要加入GCB去除����。
聲明:本文所用圖片、文字來源《食品工業(yè)科技》��,版權(quán)歸原作者所有�。如涉及作品內(nèi)容、版權(quán)等問題�����,請與本網(wǎng)聯(lián)系
相關(guān)鏈接:乙腈��,乙酸銨����,鹽酸



















登錄后才可以評論