磁固相萃取-氣相色譜法檢測黃瓜中丙溴磷(二)
發(fā)布時間:2021-10-09 14:43
編輯者:特邀作者周世紅
2 結(jié)果與分析
2.1 Fe3O4@C-MNPs的合成與表征
2.1.1 Fe3O4@C-MNPs的合成與結(jié)構(gòu)表征
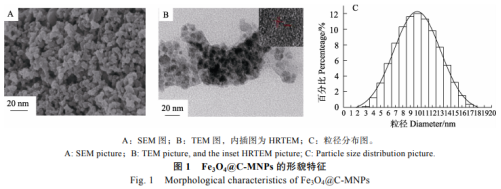
以FeCl3•6H2O�、葡萄糖、聚乙二醇���、氨水為原料采用一步水熱法在最優(yōu)條件下合成了Fe3O4@C-MNPs��。為了考察所合成材料的形貌�,通過SEM��、TEM對其進行了表征。如圖1A所示�,材料呈類似球形的顆粒結(jié)構(gòu),顆粒尺寸分布較均一�����。通過TEM(圖1B)圖可以看到明顯的碳包覆層����。這也進一步說明成功將碳包覆上Fe3O4-MNPs����。圖1B中內(nèi)插圖為Fe3O4@C-MNPs的HRTEM圖,顯示Fe3O4@C-MNPs晶格間距為0.27nm��。圖1C為通過DLS表征Fe3O4@C-MNPs的粒徑分布直方圖��,顯示其粒徑主要分布在8~12nm范圍內(nèi)�,最大粒徑分布在10nm,這與TEM和SEM顯示的結(jié)果一致�。
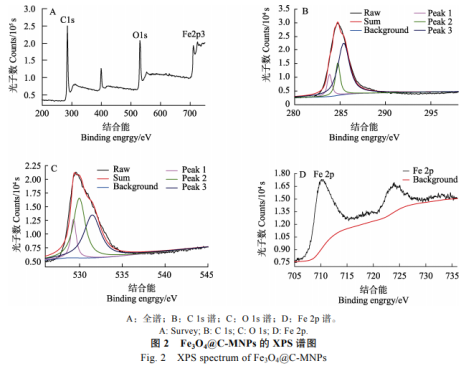
為了進一步研究Fe3O4@C-MNPs的組成結(jié)構(gòu),對該材料進行了XPS分析��,通過XPS全譜可以看到該材料主要是由C���、O����、Fe三種元素組成(圖2A)。各元素含量分別為C70.94%�����、023.11%�����、Fe5.95%����,其中高分辨C1s譜(圖2B)含3個不同結(jié)合能峰,分別為283.93�����、284.8��、285.4eV��。高分辨O1s譜(圖2C)含3個不同結(jié)合能峰����,分別在529.3�����、529.9���、531.5eV分別為混合氧化物中結(jié)合氧、-OOH和-OH峰���。高分辨Fe2p(圖2D)在710.2、723.9eV出現(xiàn)2個特征峰���。
2.1.2 Fe3O4@C-MNPs的光譜性能表征
為了進一步驗證所合成材料晶體結(jié)構(gòu)與Fe3O4磁性納米粒子一致�,對所制備的Fe3O4磁性納米顆粒和Fe3O4@C-MNPs進行了X射線衍射分析�����。如圖3A所示��,F(xiàn)e3O4@C-MNPs的XRD圖中的衍射峰2θ等于30.2°�����、35.6°����、43.4°�����、53.7°��、57.6°和62.8°分別對應(yīng)于Fe3O4的(220)�、(311)���、(400)���、(422)、(511)和(440)晶面�,其與立方晶系反尖晶石結(jié)構(gòu)標準卡片(CPDSNo.88-0866,D=6.75Å)相吻合���,衍射譜圖中未見雜峰�����,表明樣品里無雜質(zhì),且晶型單一說明所合成材料的晶體結(jié)構(gòu)與Fe3O4晶體結(jié)構(gòu)一致�。

將同樣采用水熱法合成的Fe3O4磁性納米粒子和Fe3O4@C-MNPs進行紅外光譜分析�,如圖3B為Fe3O4磁性納米粒子和Fe3O4@C-MNPs在500~4000cm-1范圍內(nèi)的吸收峰。紅外圖譜Fe3O4磁性納米粒子和Fe3O4@C-MNPs在660cm-1處都出現(xiàn)的特征峰對應(yīng)于Fe-O吸收峰����,在2900~3000cm-1區(qū)域比較弱的吸收峰為-OH的伸縮振動峰Fe3O4@C-MNPs在1601cm-1和1691cm-1的吸收峰分別對應(yīng)于C=O和C=C特征吸收峰����,而Fe3O4磁性納米粒子在此處吸收峰較弱,由此可以看出通過水熱法成功地合成了Fe3O4@C-MNPs���。
2.1.3 Fe3O4@C-MNPs的磁性能表征
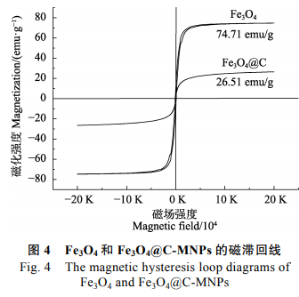
Fe3O4磁性納米粒子和Fe3O4@C-MNPs在室溫下的磁滯回線圖�。由圖4可以看出,在最大磁場強度20000Oe的外加磁場下�����,二者的矯頑力與剩磁率均接近零�,由此可知�����,制備的Fe3O4磁性納米粒子和Fe3O4@C-MNPs粒子均為超順磁性。Fe3O4磁性納米粒子的飽和磁化強度為74.71emu/g,而經(jīng)過表面修飾的Fe3O4@C-MNPs由于表面被非磁性的碳層包覆���,飽和磁化強度減小為26.51emu/g,但對于普通磁場仍保持了足夠的磁響應(yīng)���。據(jù)文獻報道�,材料飽和磁化強度大于16.3emu/g即可用于固液分離���,因此制備的Fe3O4@C-MNPs顆?���?梢杂糜趶?fù)雜樣品中殘留農(nóng)藥的富集分離。
2.2 丙溴磷的檢測
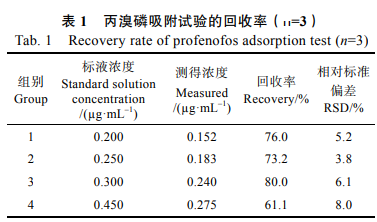
為了驗證Fe3O4@C-MNPs材料對丙溴磷的吸附效果��,按低�����、中��、高原則配制了4個不同濃度(標記為1���、2��、3�、4組)的丙溴磷標準溶液�����,分別加入30mg��,F(xiàn)e3O4@C-MNPs材料按1.2.3的方法進行處理�,計算回收率,每個濃度做3組平行,結(jié)果如表1所示����。丙溴磷吸附實驗結(jié)果表明,F(xiàn)e3O4@C-MNPs材料對于農(nóng)藥丙溴磷具有一定的吸附性����。因此后續(xù)實驗對材料吸附丙溴磷的磁固相萃取過程進行了優(yōu)化,以期獲得更高的農(nóng)藥回收率����。
2.3 丙溴磷檢測中磁固相萃取條件的優(yōu)化
2.3.1 優(yōu)化Fe3O4@C-MNPs的用量
本研究是利用Fe3O4@C-MNPs對于丙溴磷具有良好的吸附能力從而將其從復(fù)雜基質(zhì)中富集分離出來,因此Fe3O4@C-MNPs的添加量將極大的影響檢測結(jié)果���。為了考察Fe3O4@C-MNPs的最佳用量�����,在室溫下控制丙溴磷的濃度及其他條件不變��,改變材料的添加量(20�、30���、40����、50�����、60��、70mg)設(shè)計6組吸附實驗,每組平行3次�����,最終結(jié)果以丙溴磷回收率作為指標。結(jié)果如圖5A,丙溴磷的回收率隨著Fe3O4@C-MNPs的添加量呈先增大后平穩(wěn)的走勢���。在材料添加量為0.06g時達到最大回收率,回收率為87.33%~90.35%�����。該回收率在標準范圍內(nèi),說明該材料對丙溴磷吸附性良好�,可以進行后續(xù)的優(yōu)化實驗。
聲明:本文所用圖片���、文字來源《熱帶作物學報》,版權(quán)歸原作者所有�。如涉及作品內(nèi)容��、版權(quán)等問題�����,請與本網(wǎng)聯(lián)系
相關(guān)鏈接:丙溴磷,聚乙二醇,氨水


















登錄后才可以評論