薄層色譜法鑒別槐枝及高效液相色譜法測定其中蘆丁含量(二)
發(fā)布時間:2021-11-30 22:24
編輯者:特邀作者周世紅
2.2 薄層色譜鑒別結(jié)果
根據(jù)2.1薄層色譜鑒別條件,建立槐枝的薄層色譜鑒別方法:稱取槐枝粉末(過二號篩)2g����,加入石油醚(30~60℃)30mL超聲處理20min,過濾���,棄去濾液���,濾渣揮干溶劑后,加入50mL甲醇超聲處理(500W����,頻率40kHz)30min,過濾���,取濾液蒸干���,加入2mL甲醇溶解殘渣,即為供試品溶液�。按1.2.1方法制備對照品溶液。按照薄層色譜法(2020年版《中國藥典:四部》 通則 0502)試驗方法,分別吸取供試品溶液和對照品溶液各2μL��,分別點于同一硅膠G薄層板上����,以乙酸乙酯–甲醇–甲酸–水(體積比為8∶1∶1∶1)溶液為展開劑�,展開,取出��,晾干����,噴三氯化鋁試液,待乙醇揮干后�,置于紫外光燈(365nm)下檢視,結(jié)果如圖2所示��。由圖2可以看出�,供試品色譜圖在與對照品色譜圖相應(yīng)位置上,呈現(xiàn)相同顏色的熒光斑點���。

2.3 系統(tǒng)適應(yīng)性試驗
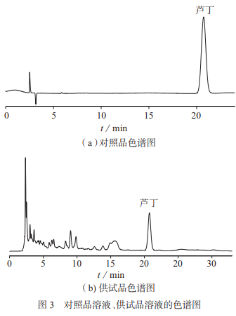
分別精密吸取1.3.2中的對照品溶液和1.3.3中的供試品溶液����,各10μL,按1.3.1色譜條件測定�����,理論板數(shù)按蘆丁色譜峰計不低于3000����,系統(tǒng)適應(yīng)性試驗符合要求。對照品溶液�、供試品溶液色譜圖如圖3所示。
2.4 蘆丁含量測定提取條件選擇
選擇編號為1#的槐枝樣品粉末���,分別選擇甲醇�����、70%乙醇溶液��、50%甲醇溶液及50%乙醇溶液4種提取溶劑對蘆丁進行提取����,考察不同提取溶劑的提取效果�。結(jié)果發(fā)現(xiàn)以50%甲醇溶液為提取溶劑時,色譜峰面積最大����,基線平穩(wěn)���,故采用50%甲醇為提取溶劑。以50%甲醇溶液為提取溶劑��,分別采用超聲法(30��、60min)����、回流法(30�����、60min)對蘆丁進行提取�,考察不同提取方法的提取效果。結(jié)果發(fā)現(xiàn)超聲提?����。?0���、60min)及回流提?。?0、60min)的色譜峰面積無明顯差異�,故選用超聲法(30min)對樣品進行提取。
2.5 色譜條件優(yōu)化
分別選用瑞典Nouryon的Kromasil100–5–C18(250mm×4.6mm��,5.0μm)色譜柱�、島津企業(yè)管理(中國)有限公司的VP–ODSC18(250mm×4.6mm,5.0μm)色譜柱及美國安捷倫科技有限公司的AgilentZorbaxSBC18(250mm×4.6mm�,5.0μm)色譜柱進行試驗,發(fā)現(xiàn)不同色譜柱之間無明顯差異���,均能實現(xiàn)色譜分離�,理論板數(shù)�����,分離度及拖尾因子均符合《中國藥典》2020年版的規(guī)定���。本實驗選用瑞典Nouryon的Kromasil100–5–C18(250mm×4.6mm�����,5.0μm)色譜柱作為分離柱��。
在1.3.1色譜條件下�,分別考察了甲醇–0.01mol/L乙酸銨(體積比為47∶53)溶液、甲醇–1%冰醋酸(體積比為32∶68)溶液��、乙腈–2.5%冰醋酸(體積比為13∶87)溶液3種流動相系統(tǒng)�。結(jié)果發(fā)現(xiàn)采用乙腈–2.5%冰醋酸溶液作為流動相時,色譜圖中的溶劑峰明顯�;采用甲醇–0.01mol/L乙酸銨溶液、甲醇–1%冰醋酸溶液作為流動相時�,蘆丁的色譜峰尖銳,理論板數(shù)高�,分離度良好。由于甲醇–0.01mol/L乙酸銨溶液配制較復(fù)雜����,使用鹽溶液操作不當容易阻塞色譜柱����,損壞高效液相色譜儀,而甲醇–1%冰醋酸溶液配制簡單�,操作容易。綜合考慮�,選擇甲醇–1%冰醋酸(體積比為32∶68)溶液為流動相。
2.6 線性方程與檢出限
自動進樣器分別精密吸取1.3.2中的對照品溶液3�����、7、10�、13、15μL注入液相色譜儀��,在1.3.1色譜條件下�,依次進樣測定,以蘆丁的質(zhì)量為橫坐標�����,以色譜峰面積為縱坐標繪制標準曲線���,計算線性方程和相關(guān)系數(shù)����。
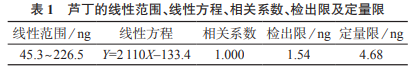
用甲醇將對照品溶液稀釋10倍���,然后逐級稀釋進樣分析�,以3倍信號和噪音的比值(S/N)對應(yīng)的質(zhì)量作為方法檢出限�����,5倍信噪比對應(yīng)的質(zhì)量作為定量限����。蘆丁的線性范圍�����、線性方程�����、相關(guān)系數(shù)����、檢出限及定量限見表1����。
2.7 穩(wěn)定性試驗
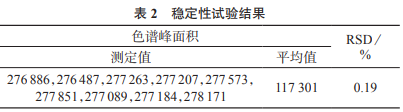
選擇1號槐枝樣品粉末(過二號篩)�����,精密稱取約0.5g����,按照1.3.3方法制備供試品溶液,在1.3.1色譜條件下��,分別于制備完成后第0、1���、2�����、3��、5��、7�、10��、12����、24h進樣測定,結(jié)果見表2���。由表2可知���,在24h內(nèi)色譜峰面積測定結(jié)果的相對標準偏差為0.19%,表明樣品溶液在24h內(nèi)穩(wěn)定性較好�����。
2.8 精密度試驗
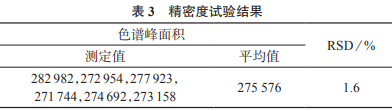
取1號槐枝樣品粉末(過二號篩)6份,每份約0.5g����,精密稱定,按照1.3.3方法制備供試品溶液�,在1.3.1色譜條件下分別進樣測定,結(jié)果見表3����。由表3可知,色譜峰面積測定結(jié)果的相對標準偏差為1.6%��,表明該方法精密度較好���,滿足測定要求�。
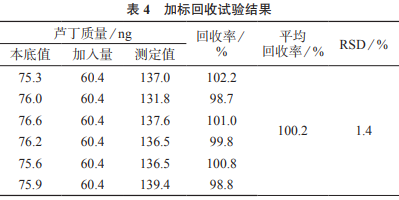
2.9 加標回收試驗
精密稱取1號槐枝樣品粉末(過二號篩)6份�����,每份約0.25g��,分別加入質(zhì)量濃度為7.54μg/mL的蘆丁對照品溶液20mL����,再精密加入25mL水和5mL甲醇,密塞��,稱量并記錄溶液質(zhì)量���,超聲處理(500W���,頻率40kHz)30min,冷卻���,再次稱量溶液質(zhì)量����,用50%甲醇溶液補足減失的質(zhì)量��,搖勻���,過濾����,取續(xù)濾液,在1.3.1色譜條件下分別進樣測定�����,結(jié)果見表4�����。由表4可知��,樣品加標回收率為98.7%~102.2%�,測定結(jié)果的相對標準偏差為1.4%,表明該方法準確度較高�����。
2.1 實際樣品測定
分別精密稱取1~10號槐枝樣品粉末(過二號篩)��,各約0.5g����,按1.3.3方法制備供試品溶液,在1.3.1色譜條件下分別進樣測定�,結(jié)果見表5。

由表5可知�����,10批槐枝樣品中蘆丁的質(zhì)量分數(shù)在0.10%~0.16%之間�。不同槐枝樣品之間蘆丁含量差異較大,可能是在槐枝采集過程中���,存在以老枝摻入嫩枝入藥的情形���,而老枝中蘆丁的含量較低。
3 結(jié)語
樣品先加石油醚(30~60℃)脫色�,然后用甲醇進行提取,以乙酸乙酯–甲醇–甲酸–水(體積比為8∶1∶1∶1)為展開劑���,顯色后斑點清晰���,分離度好,可用于槐枝的定性鑒別�����。采用50%甲醇作為提取溶劑�����,以甲醇–1%冰醋酸溶液(體積比為32∶68)為流動相,建立了高效液相色譜法測定槐枝中蘆丁含量的方法�����。該方法簡便易行�、準確可靠,可為槐枝質(zhì)量控制提供技術(shù)支持�����。
相關(guān)鏈接:乙酸乙酯�����,石油醚����,蘆丁,冰醋酸
聲明:本文所用圖片����、文字來源《化學(xué)分析計量》,版權(quán)歸原作者所有。如涉及作品內(nèi)容、版權(quán)等問題�,請與本網(wǎng)聯(lián)系



















登錄后才可以評論