同位素稀釋氣相色譜-串聯(lián)質(zhì)譜法測定動物源食品中克霉唑殘留量(二)
發(fā)布時間:2021-06-17 16:59
編輯者:特邀作者周世紅
1.2.7色譜條件
HP-5MS色譜柱(30m×0.25mm×0.25μm)���,程序升溫:初溫60℃��,保持1min�����,以30℃/min升至200℃��,冉以15℃/mjn升至300℃����,保持5min。進(jìn)樣口溫度240℃�,不分流進(jìn)樣,進(jìn)樣量1μL��,載氣為氦氣(純度≥99.999%)�,流速1.0mL/min。
1.2.8質(zhì)潛條件
電子電離源(EI)�,電離能量70eV,離子源溫度280℃�,傳輸線溫度300℃,多反應(yīng)監(jiān)測(MRM)模式����,具體條件見表1�。

1.3數(shù)據(jù)處理
采用Tracefinder3.0軟件處理數(shù)據(jù)�。
2結(jié)果與分析
2.1質(zhì)譜參數(shù)的優(yōu)化
在EI電離源下,采用SCAN模式�,對5.0mg/L的克霉嘩及克霉唑-d5分別進(jìn)行一級質(zhì)譜全掃描分析,選擇豐度最高的作為母離子��。在不同碰撞能量下�����,對目標(biāo)物的母離子分別進(jìn)行二級質(zhì)譜掃描����,得到每個母離子的碎片離子,選擇豐度較高的3個子離子�,配制不同種類的空白樣品基質(zhì)標(biāo)準(zhǔn)溶液���,在MRM模式下優(yōu)化各種質(zhì)譜條件����,剔除易受雜質(zhì)干擾�����,選擇半度最高的2個子離子作為定量離子和定性離子,最后再對每對離子對的碰撞能做優(yōu)化以達(dá)到最佳靈敏度�����。標(biāo)準(zhǔn)溶液MRM色譜圖見圖1�。
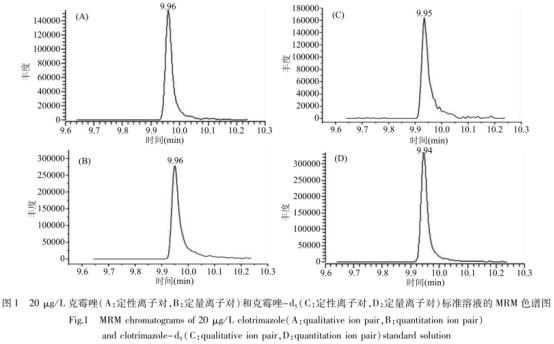
2.2前處理條件的優(yōu)化
2.2.1提取
實驗中發(fā)現(xiàn)使用常規(guī)的高速均質(zhì)提取豬肥肉(豬脂肪)和豬肉(同時含豬肥肉和精肉)時,由于樣品粘在均質(zhì)刀頭上��,無法充分和提取溶劑接觸�,導(dǎo)致克霉嘩的提取效率不高。本實驗首先將樣品在碾缽中與硅藻土充分混勻研磨��,硅藻土顆粒不僅能吸收樣品中的水分���,且能和樣品的肌肉或脂肪組織充分接觸���,使后續(xù)ASE提取時有機(jī)溶劑能充分滲透到樣品中,大大提高了克霉唑的提取效率��?����?紤]到與后續(xù)GPC凈化流動相的匹配,本實驗選擇了乙酸乙酯/環(huán)己烷(體積比1:1)為提取溶劑����。
2.2.2凈化
動物源食品或多或少含有一定量的脂肪,去除脂肪是前處理必須解決的問題���。凝膠滲透色譜是去除脂肪的有力工具���,但是實驗中發(fā)現(xiàn)通過GPC無法完全去除脂肪,否則會造成部分克霉唑損失回收率偏低�,故在GPC之后還需要通過分配步驟使殘余脂肪和克霉嘩分離。GPC洗脫液氮氣吹干后在離心管底有脂肪析出����,還需將收集到的洗脫液于45℃的水浴中氮氣吹至近干,取0.5mL乙腈飽和的正己烷渦旋振蕩溶解殘余物��,再加入0.5mL正己烷飽和的乙腈�����,渦旋振蕩并靜置分層�����,吸出乙腈層于2mL進(jìn)樣小瓶中��,向刻度離心管中重復(fù)加入0.5mL正己烷飽和的乙腈重復(fù)操作一次�����,合并乙腈層���,過0.22μm濾膜于GC-MS/MS的進(jìn)樣小瓶中�,制得待測樣品溶液���,供GC-MS/MS測定的步驟通過液液分配使脂肪和克霉唑分離�,使用0.5mL正己烷(乙腈飽和)溶解殘渣���,再用0.5mL乙腈(正己烷飽和)反提取克霉嘩��,試驗中共用0.5mL乙腈(正己烷飽和)重復(fù)提取3次����,考察每次乙腈層中克霉唑的分布��,結(jié)果發(fā)現(xiàn)用2次提取即能達(dá)到90%的回收率�,故采用0.5mL乙腈(正己烷飽和)提取2次��,合并乙腈層供GCS-MS/MS測定��。
2.3方法學(xué)驗證
2.3.1線性關(guān)系與定量限
采用內(nèi)標(biāo)法對克霉唑標(biāo)準(zhǔn)品10mg�,用乙腈溶解并轉(zhuǎn)移至100mL容量瓶中����,用乙腈定容,制得100μg/mL克霉唑標(biāo)準(zhǔn)儲備液�;取克霉嘩-d5標(biāo)準(zhǔn)品1mg,用乙腈溶解并轉(zhuǎn)移至10mL容量瓶中����,用乙腈定容,制得100μg/mL克霉嘩-d5標(biāo)準(zhǔn)儲備液�。用乙腈稀釋配制濃度分別為1、2���、5�、20��、50���、100μg/L的系列標(biāo)準(zhǔn)工作溶液����,其巾克霉嘩-d5同位素內(nèi)標(biāo)溶液濃度均為20μg/L���。制備的1�、2����、5、20��、50���、100μg/L系列基質(zhì)標(biāo)準(zhǔn)工作溶液進(jìn)行上機(jī)測定�,以克霉嘩與克霉嘩-d5的峰面積比值為縱坐標(biāo)(Y)����,克霉嘩的質(zhì)量濃度為橫坐標(biāo)(X,μg/L)繪制標(biāo)準(zhǔn)曲線��。結(jié)果表明��,在1�、2����、5���、20�、50�����、100μg/L濃度范圍內(nèi)��,克霉唑線性關(guān)系良好��,回歸方程為Y=0.0189426+0.0474255X��,決定系數(shù)(R2)為0.9995��。定量限(LOQ)采用在空白樣品中逐級降低加標(biāo)濃度的方法來確定����,得到克霉唑的定量限(LOQ,S/N≥10)為2.0μg/kg����。
2.3.2回收率與精密度
在不同動物源食品的空白樣品中�����,分別進(jìn)行2.0、5.0���、20.0μg/kg三個不同濃度水平的加標(biāo)回收實驗�����,每個水平重復(fù)測定6次����,通過同位素內(nèi)標(biāo)法計算其回收率和標(biāo)準(zhǔn)偏差(RSD)�����,結(jié)果見表2����。采用了同位素內(nèi)標(biāo)定量技術(shù),校正了目標(biāo)物的損失�����、提高了測量結(jié)果的穩(wěn)定性和重復(fù)性,測得克霉唑的回收率在82.6%~116.5%之問����,相對標(biāo)準(zhǔn)偏差為5.2%~13.5%,說明該方法的準(zhǔn)確度和精密度良好�。代表性的樣品加標(biāo)色譜圖見圖2,在克霉唑出峰處沒有雜質(zhì)干擾�����,說明本研究采用的凈化步驟取得了良好的效果�。

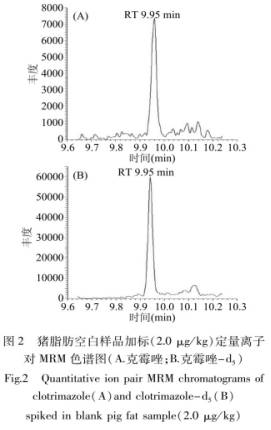
2.4實際樣品檢測
在農(nóng)貿(mào)市場和超市購買不同的動物源食品,豬肉��、雞肉����、豬脂肪、豬心��、豬肝��、豬腎��,每種樣品各3份,采用本方法進(jìn)行檢測��,結(jié)果顯示���,所有的樣品中均未檢出克霉唑藥物殘留���。
3結(jié)論
本文建立了動物源食品巾克霉嘩殘留量的同位素稀釋氣相色譜-串聯(lián)質(zhì)譜測定方法。樣品經(jīng)加速溶劑提取后�,用凝膠滲透色譜凈化結(jié)合正己烷去除樣品中的脂肪����,同位素內(nèi)標(biāo)定景,校正了目標(biāo)物的損失����、提高了測量結(jié)果的穩(wěn)定性和重復(fù)性,保證了方法有較好的回收率和精密度�����。該方法靈敏度高�、準(zhǔn)確性好,定量限為2.0μg/kg�����,同收率在82.6%~116.5%之間,相對標(biāo)準(zhǔn)偏差為5.2%~13.5%���,能夠滿足動物源食品中克霉唑殘留量的確證和定量分析���,為保障食品安全提供技術(shù)支持。
聲明:本文所用圖片�、文字來源《食品工業(yè)科技》,版權(quán)歸原作者所有���。如涉及作品內(nèi)容�����、版權(quán)等問題����,請與本網(wǎng)聯(lián)系
相關(guān)鏈接:克霉唑��,乙酸乙酯�����,乙腈

















登錄后才可以評論