高效液相色譜-串聯(lián)質(zhì)譜法同時測定葡萄酒中甜味劑����、防腐劑和色素(二)
發(fā)布時間:2021-02-24 18:33
編輯者:周世紅
2、色譜和質(zhì)譜條件的優(yōu)化
(1)流動相的選擇
反相色譜的流動相通常由水和有機(jī)溶劑(如甲醇���、乙腈)等組成�����,由于某些被測物極性較強(qiáng)��,流動相的洗脫能力不宜過強(qiáng)�,同時待分離的16種添加劑有些極性相近,保留時間相近���,采用乙腈為有機(jī)相時導(dǎo)致幾個組分“共流出”情況較甲醇嚴(yán)重����,而甲醇可使16種添加劑的保留時間相對延遲��,同時甲醇可以提高離子化程度�,峰面積比用乙腈為流動相時大�����,故選擇甲醇為實驗的有機(jī)相���;又由于流動相要進(jìn)入質(zhì)譜儀�,添加一定量的緩沖溶液可以增加響應(yīng)值�����,所以考慮甲醇-甲酸水溶液�、甲醇-乙酸銨溶液、甲醇-乙酸水溶液3種流動相體系。
以被測物在色譜柱上的分離度����、峰形、靈敏度等為考察指標(biāo)對3種流動相體系進(jìn)行了比較�����。結(jié)果表明�����,體系中添加了乙酸銨不僅更有利于分離物質(zhì)在色譜柱上的保留����,而且能夠提高色素類物質(zhì)的電離化程度,增加信號響應(yīng)�,從各物質(zhì)分離度、峰形�、響應(yīng)值以及保留時間的穩(wěn)定性等指標(biāo)進(jìn)行綜合衡量,甲醇-10mmol/L乙酸銨作為流動相時���,優(yōu)于其余兩種流動相�,故實驗選用甲醇-10mmol/L乙酸銨為流動相進(jìn)行梯度洗脫��。
(2)柱溫的選擇
柱溫能夠影響色譜柱的柱效、選擇性�����、靈敏度和穩(wěn)定性�,柱溫的改變直接影響分離效果及分離速率。升高柱溫����,色譜柱內(nèi)離子交換速率隨之升高,有利于提高柱效���、縮短分析時間。本實驗考察了柱溫分別為30℃�����、35℃�、40℃時,16種添加劑保留時間及響應(yīng)值的差別����,發(fā)現(xiàn)當(dāng)柱溫為30℃時���,16種添加劑有13種出峰時間均集中在3~4min����,分離度差���;提高柱溫至35℃時�����,柱效提高���,分離度和靈敏度均有改善�����,繼續(xù)提高柱溫�����,分離情況無明顯變化?����?紤]到色譜柱在相對較低溫度下使用壽命較長���,本實驗設(shè)定柱溫為35℃��。優(yōu)化條件下16種添加劑的MRM質(zhì)譜圖見圖1�。
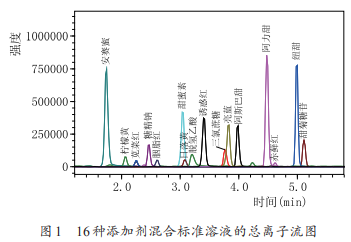
(3)質(zhì)譜條件的選擇
為獲得最佳的靈敏度和分離效果��,根據(jù)16種添加劑的分子結(jié)構(gòu)特征�,在正�����、負(fù)兩種電離模式下優(yōu)化被測物的母離子和特征子離子以及相應(yīng)的質(zhì)譜參數(shù)����。取質(zhì)量濃度為1mg/L的16種化合物單標(biāo)準(zhǔn)溶液依次采用不接色譜柱直接進(jìn)樣方式進(jìn)行質(zhì)譜全掃描檢測���,得到目標(biāo)分析物一級質(zhì)譜圖�,再用氬氣轟擊該母離子���,得到其二級質(zhì)譜圖�����,利用儀器的自動優(yōu)化功能����,分別對Q1���、Q3、CE等進(jìn)行優(yōu)化���,確定16種添加劑的母離子和子離子的最佳質(zhì)譜條件�,以強(qiáng)度較大的子離子作為定量離子��,強(qiáng)度稍小的子離子為定性離子����。
3�、方法學(xué)評價
(1)方法的線性范圍和檢出限
將含16種被測物的混合標(biāo)準(zhǔn)儲備液添加至稀釋5倍的空白葡萄酒樣品中,分別配制系列混合標(biāo)準(zhǔn)工作液���。
色譜柱:AcquityUPLCHSST3柱(100mm×2.1mm,1.8µm,美國Waters公司)����。流動相:(A)甲醇和(B)10mmol/L乙酸銨水溶液����,流速0.35mL/min���;梯度洗脫程序為:0min,甲醇的體積分?jǐn)?shù)為10%��;0~3min�����,甲醇的體積分?jǐn)?shù)從10%升至90%���,并保持2.5min�����,5.5~5.51min;甲醇的體積分?jǐn)?shù)從90%降至10%����;5.51min~6.5min,甲醇的體積分?jǐn)?shù)保持10%����。柱溫為35℃;進(jìn)樣體積:2μL��。
分析儀器:LCMS-8050;離子源:電噴霧離子源(ESI)���;掃描方式:正負(fù)離子同時掃描���;離子源接口電壓:0.5kV;霧化氣:氮氣3.0L/min���;干燥氣:氮氣10L/min����;碰撞氣:氬氣;DL溫度:250℃�����;加熱模塊溫度:400℃�;掃描模式:多反應(yīng)監(jiān)測(MRM);駐留時間:15ms�;延遲時間:3ms��;以各組分定量離子色譜峰面積對相應(yīng)的質(zhì)量濃度繪制各被測物的標(biāo)準(zhǔn)工作曲線���,結(jié)果表明,線性關(guān)系良好���,相關(guān)系數(shù)(r2)都在0.995以上�����。
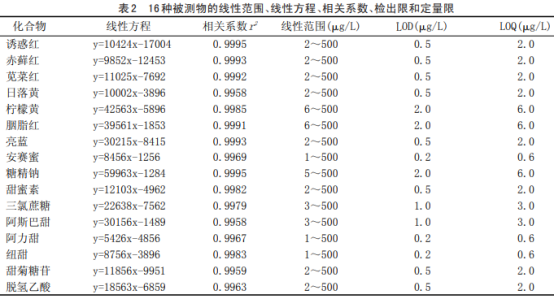
用葡萄酒樣品低加標(biāo)水平計算各組分的檢出限和定量限���,用信噪比為3確定方法的檢出限(LOD)�����,用信噪比為10確定方法的定量限(LOQ)�����,各組分在葡萄酒樣品中的檢出限范圍在0.2~2μg/L之間����。結(jié)果見表2。
(2)精密度和回收率
用微量移液器向空白葡萄酒樣品中準(zhǔn)確加入一定量的16種待測物混合標(biāo)準(zhǔn)溶液��,配成低��、中、高3個濃度水平進(jìn)行回收率實驗�����。分別準(zhǔn)確稱取16種添加劑標(biāo)準(zhǔn)品于10mL棕色容量瓶中,用水或甲醇溶解并定容�����,配制成質(zhì)量濃度為1mg/mL的單標(biāo)儲備液����,于-18℃下避光保存��。用甲醇稀釋并配制中間濃度的標(biāo)準(zhǔn)工作液��,于4℃避光保存����。
根據(jù)需要用流動相逐級稀釋,配制成適當(dāng)濃度的混合標(biāo)準(zhǔn)工作液���,現(xiàn)配現(xiàn)用����。取葡萄酒樣品2.0mL于10mL容量瓶中�,用超純水定容��,超聲混勻�,過0.22μm濾膜��,濾液供測定���。
色譜柱:AcquityUPLCHSST3柱(100mm×2.1mm,1.8µm����,美國Waters公司)��。
流動相:(A)甲醇和(B)10mmol/L乙酸銨水溶液����,流速0.35mL/min;梯度洗脫程序為:0min�,甲醇的體積分?jǐn)?shù)為10%;0~3min��,甲醇的體積分?jǐn)?shù)從10%升至90%�,并保持2.5min,5.5~5.51min���;甲醇的體積分?jǐn)?shù)從90%降至10%���;5.51min~6.5min�,甲醇的體積分?jǐn)?shù)保持10%�。
柱溫為35℃;進(jìn)樣體積:2μL�����。
分析儀器:LCMS-8050���;離子源:電噴霧離子源(ESI);掃描方式:正負(fù)離子同時掃描���;離子源接口電壓:0.5kV�����;霧化氣:氮氣3.0L/min�����;干燥氣:氮氣10L/min����;碰撞氣:氬氣;DL溫度:250℃����;加熱模塊溫度:400℃;掃描模式:多反應(yīng)監(jiān)測(MRM)���;駐留時間:15ms�;延遲時間:3ms���。
進(jìn)行6次重復(fù)實驗�����,計算其回收率及相對標(biāo)準(zhǔn)偏差(RSD)�����。結(jié)果表明��,各目標(biāo)物平均回收率在89.1%~106.4%之間����,相對標(biāo)準(zhǔn)偏差小于9.7%,方法的準(zhǔn)確度和精密度均符合多殘留分析的要求�����。
4��、實際樣品的測定
按所建立的方法對采購的10批次葡萄酒樣品進(jìn)行了16種添加劑的篩查��,每個樣品重復(fù)測定3次�����。所檢葡萄酒樣品中有1個檢出糖精鈉和甜蜜素�,含量分別為26.3μg/L、38.9μg/L��,其余樣品的檢測結(jié)果均為陰性��。陽性樣品的MRM質(zhì)譜圖見圖2�。

三�、結(jié)論
本研究建立了高效液相色譜-串聯(lián)質(zhì)譜儀同時測定葡萄酒中甜味劑、防腐劑和色素含量的方法����,前處理簡單、分析速度快�����、準(zhǔn)確度和靈敏度高。實際樣品的檢測表明���,該方法能夠滿足葡萄酒甜味劑�����、防腐劑和色素殘留的分析要求����,與國標(biāo)方法相比�����,大大提高了分析效率和降低了分析成本��,能夠滿足葡萄酒中痕量分析要求�����,非常適合大批量葡萄酒樣品中16種添加劑的定性和定量分析����。
聲明:本文所用圖片�、文字來源《中國食品添加劑》�����,版權(quán)歸原作者所有�。如涉及作品內(nèi)容、版權(quán)等問題���,請與本網(wǎng)聯(lián)系
相關(guān)鏈接:色譜��,甲醇���,超純水,甜味劑
















登錄后才可以評論